Paired integration and query-to-reference mapping#
In this tutorial, we demonstrate how to use Multigrate’s integration module for paired integration, i.e. reference building, and mapping on the query onto the built reference. We will use NeurIPS 2021 CITE-seq dataset [LBC+21].
import sys
# if branch is stable, will install via pypi, else will install from source
branch = "latest"
IN_COLAB = "google.colab" in sys.modules
if IN_COLAB and branch == "stable":
!pip install multigrate[tutorials]
elif IN_COLAB and branch != "stable":
!pip install muon
!pip install --quiet --upgrade jsonschema
!pip install git+https://github.com/theislab/multigrate
import anndata as ad
import multigrate as mtg
import muon
import scanpy as sc
import scvi
import warnings
warnings.filterwarnings("ignore")
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.4.0.post1
Data loading#
The data contains 90,261 bone marrow mononuclear cells. This CITE-seq (i.e. paired gene expression and surface protein adundance) dataset was generated at 4 different sites introducing some batch effect. After the quality control performed by the authors, the data contains measurements from 13,953 genes and 134 proteins.
data_path = "GSE194122_openproblems_neurips2021_cite_BMMC_processed.h5ad"
try:
adata = sc.read_h5ad(data_path)
except OSError:
!wget 'ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE194nnn/GSE194122/suppl/GSE194122_openproblems_neurips2021_cite_BMMC_processed.h5ad.gz'
!gzip -d GSE194122_openproblems_neurips2021_cite_BMMC_processed.h5ad.gz
adata = sc.read_h5ad(data_path)
adata
AnnData object with n_obs × n_vars = 90261 × 14087
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id'
uns: 'dataset_id', 'genome', 'organism'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
Data preparation#
We subsample the data to speed up the training.
sc.pp.subsample(adata, n_obs=20000)
adata
AnnData object with n_obs × n_vars = 20000 × 14087
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id'
uns: 'dataset_id', 'genome', 'organism'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
We split the data into the gene counts and protein counts.
rna = adata[:, adata.var["feature_types"] == "GEX"].copy()
rna
AnnData object with n_obs × n_vars = 20000 × 13953
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id'
uns: 'dataset_id', 'genome', 'organism'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
adt = adata[:, adata.var["feature_types"] == "ADT"].copy()
adt
AnnData object with n_obs × n_vars = 20000 × 134
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id'
uns: 'dataset_id', 'genome', 'organism'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
# to free memory
del adata
RNA preprocessing#
Next, we log-normalize the raw counts and subset the RNA data to 2,000 highly variable genes.
rna.X = rna.layers["counts"].copy()
sc.pp.normalize_total(rna, target_sum=1e4)
sc.pp.log1p(rna)
n_top_genes = 2000
batch_key = "Site"
sc.pp.highly_variable_genes(rna, n_top_genes=n_top_genes, batch_key=batch_key, subset=True)
rna
AnnData object with n_obs × n_vars = 20000 × 2000
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'dataset_id', 'genome', 'organism', 'log1p', 'hvg'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
ADT preprocessing#
We will use centered log-ratio (CLR) normalization for the ADT modality.
adt.X = adt.layers["counts"].copy()
muon.prot.pp.clr(adt)
adt.layers["clr"] = adt.X.copy()
Data setup#
Next, we create one AnnData object from the RNA and ADT objects and specify which layers to use during training.
adata = mtg.data.organize_multimodal_anndatas(
adatas=[[rna], [adt]], # a list of anndata objects per modality, RNA-seq always goes first
layers=[["counts"], ["clr"]], # if need to use data from .layers, if None use .X
)
adata
AnnData object with n_obs × n_vars = 20000 × 2134
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train', 'group'
var: 'modality'
uns: 'modality_lengths'
layers: 'counts'
The data comes from 4 different sites, so we select one of the sites as the query.
query = adata[adata.obs[batch_key] == "site1"].copy()
adata = adata[adata.obs[batch_key] != "site1"].copy()
(adata, query)
(AnnData object with n_obs × n_vars = 16433 × 2134
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train', 'group'
var: 'modality'
uns: 'modality_lengths'
layers: 'counts',
AnnData object with n_obs × n_vars = 3567 × 2134
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train', 'group'
var: 'modality'
uns: 'modality_lengths'
layers: 'counts')
We specify which covariates we want to integrate out during training, i.e. if there is batch effect in the data we want to correct, which covariate should be used as the batch covariate. Please refer to the API if you need to specify multiple covariates or continuous covariates.
rna_indices_end = rna.shape[1]
mtg.model.MultiVAE.setup_anndata(
adata,
categorical_covariate_keys=[batch_key],
rna_indices_end=rna_indices_end,
)
adata
AnnData object with n_obs × n_vars = 16433 × 2134
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train', 'group', 'size_factors', '_scvi_batch'
var: 'modality'
uns: 'modality_lengths', '_scvi_uuid', '_scvi_manager_uuid'
obsm: '_scvi_extra_categorical_covs'
layers: 'counts'
Model setup and training#
In the next step, we initialize the model. We need to pass which adata the model will be trained on and which losses should be used for each of the modalities. Since we use raw counts for RNA, we will use negative binomial loss here ('nb'), and MSE ('mse') for the normalized ADT counts.
In general, we recommend the following distributions/losses for different modalities:
Modality |
Distribution |
Loss name |
Data normalization |
|---|---|---|---|
Gene expression |
Negative binomial |
|
None |
Protein abundance |
Normal |
|
Centered log ratio |
Chromatin accesibility |
Normal |
|
Total-counts + log-normalization |
Mass spectrometry |
Normal |
|
Total-counts + log-normalization |
Custom |
Normal |
|
Total-counts + log-normalization or any |
vae = mtg.model.MultiVAE(
adata,
losses=["nb", "mse"],
)
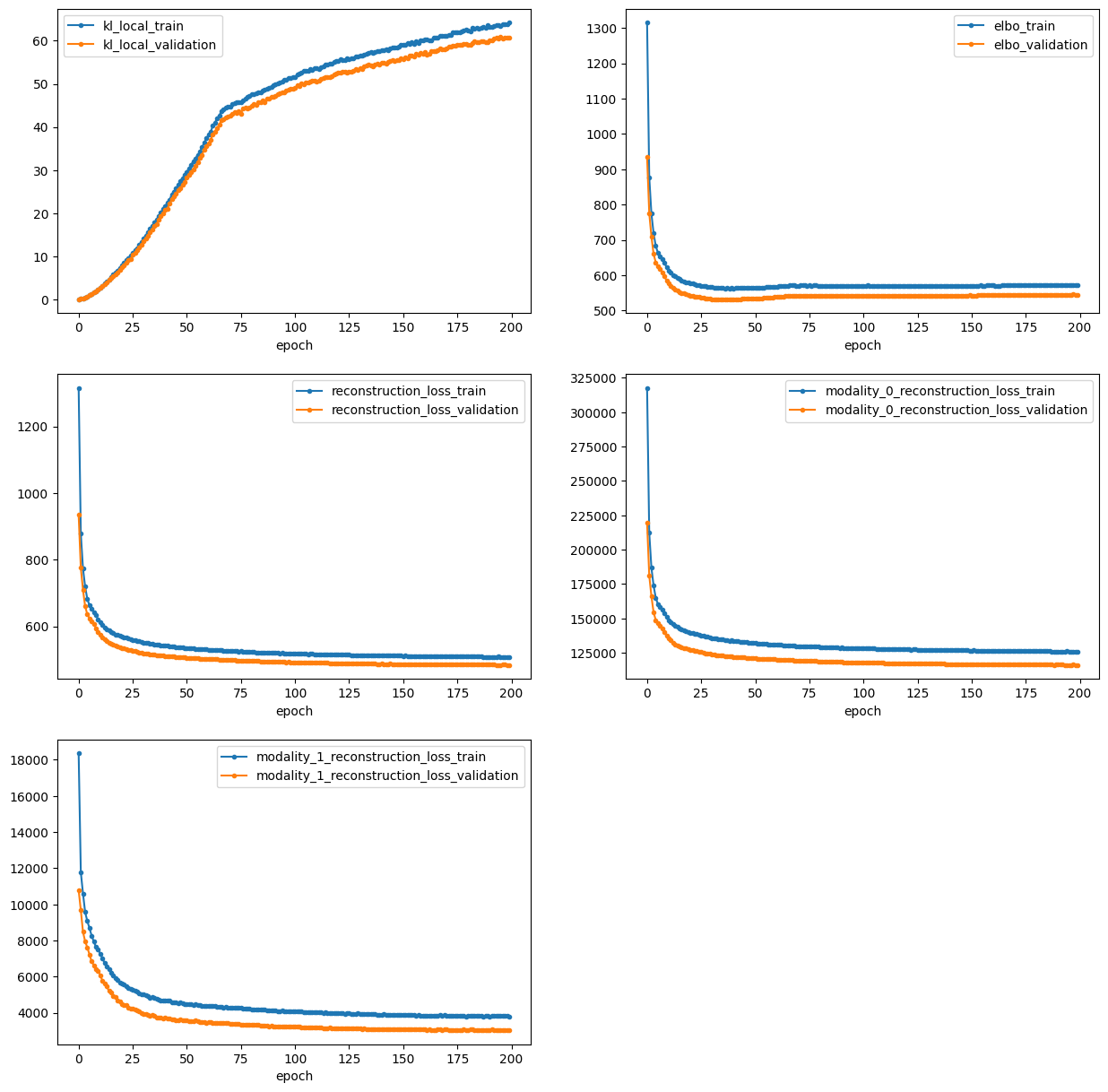
Now, we finally are set and can train the model and visualize the losses.
vae.train()
vae.plot_losses()
Visualizing the latent space#
Next, we retrieve the learned latent representation; it is automatically saved in adata.obsm['X_multigrate'].
vae.get_model_output()
adata
AnnData object with n_obs × n_vars = 16433 × 2134
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train', 'group', 'size_factors', '_scvi_batch'
var: 'modality'
uns: 'modality_lengths', '_scvi_uuid', '_scvi_manager_uuid'
obsm: '_scvi_extra_categorical_covs', 'X_multigrate'
layers: 'counts'
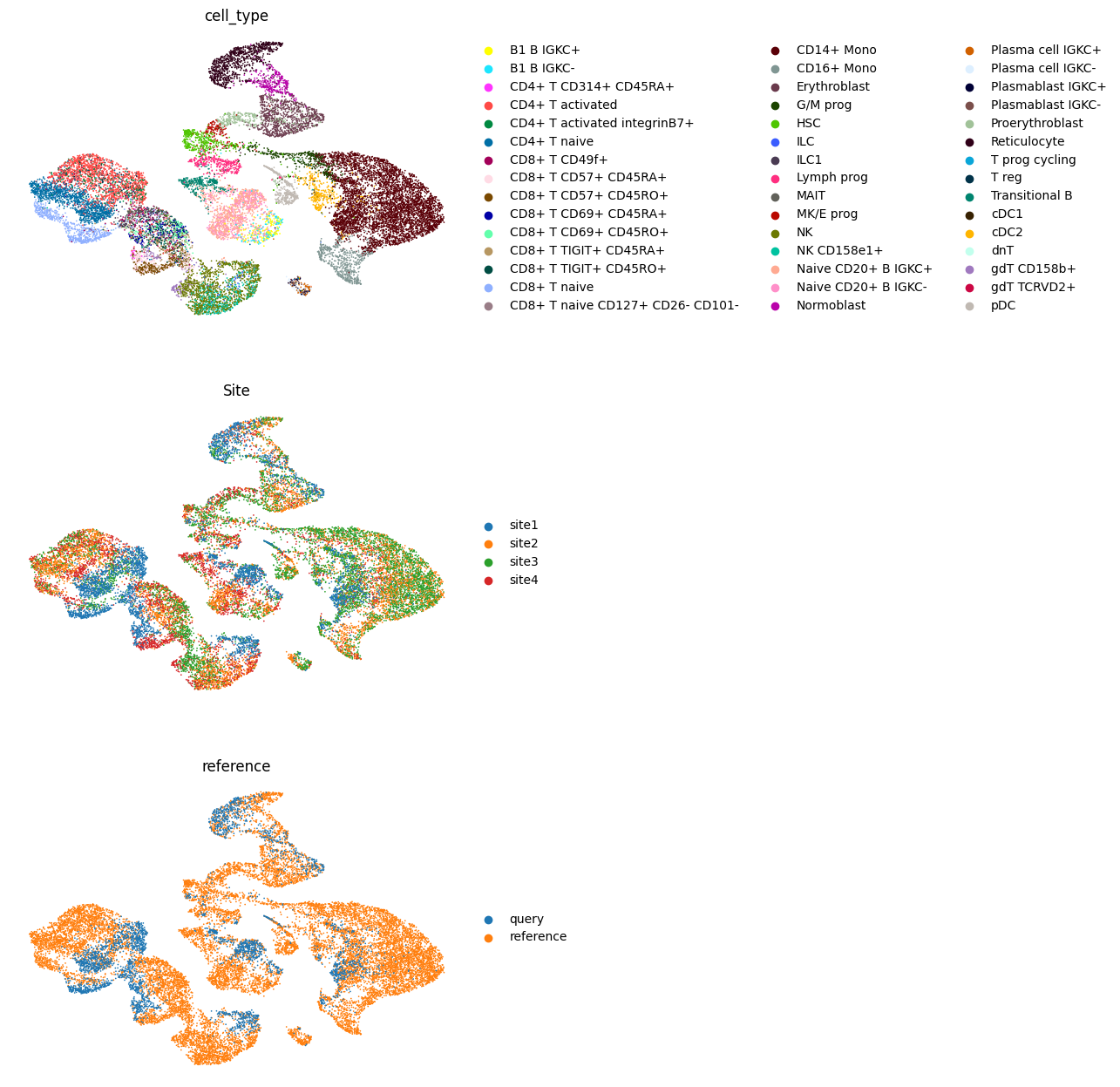
Finally, we visualize the latent space.
sc.pp.neighbors(adata, use_rep="X_multigrate")
sc.tl.umap(adata)
sc.pl.umap(adata, color=["cell_type", "Site"], frameon=False, ncols=1)

Preparing the query#
Multigrate is equipped with scArches approach [LNL+22] to map new query data onto existing references.
First, we need to update the model architecture to add weights for the new query batches.
new_vae = mtg.model.MultiVAE.load_query_data(query, reference_model=vae)
Next, we fine-tune the newly added weights to optimize the reconstruction of the query data. We set weight_decay to zero to make sure that the rest of the weights in the model will not be changed.
new_vae.train(weight_decay=0)
Now, we obtain the latent representation of the query and visualize both the reference and the query together.
new_vae.get_model_output(query)
query
AnnData object with n_obs × n_vars = 3567 × 2134
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train', 'group', 'size_factors', '_scvi_batch'
var: 'modality'
uns: 'modality_lengths', '_scvi_uuid', '_scvi_manager_uuid'
obsm: '_scvi_extra_categorical_covs', 'X_multigrate'
layers: 'counts'
adata.obs["reference"] = "reference"
query.obs["reference"] = "query"
adata_both = ad.concat([adata, query])
adata_both
AnnData object with n_obs × n_vars = 20000 × 2134
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train', 'group', 'size_factors', '_scvi_batch', 'reference'
obsm: '_scvi_extra_categorical_covs', 'X_multigrate'
layers: 'counts'
sc.pp.neighbors(adata_both, use_rep="X_multigrate")
sc.tl.umap(adata_both)
sc.pl.umap(adata_both, color=["cell_type", "Site", "reference"], ncols=1, frameon=False)