Trimodal integration and query-to-reference mapping#
In this notebook we demonstrate how to build a trimodal reference atlas and map unimodal as well as multimodal query data onto the reference. We use publically available dataset from NeurIPS 2021 workshop [LBC+21]. Please also refer to the tutorial on paired integration for more detailed explanation of the preprocessing steps and the model setup.
import sys
# if branch is stable, will install via pypi, else will install from source
branch = "latest"
IN_COLAB = "google.colab" in sys.modules
if IN_COLAB and branch == "stable":
!pip install multigrate[tutorials]
elif IN_COLAB and branch != "stable":
!pip install muon
!pip install --quiet --upgrade jsonschema
!pip install git+https://github.com/theislab/multigrate
import anndata as ad
import json
import multigrate as mtg
import numpy as np
import scanpy as sc
import scvi
import warnings
warnings.filterwarnings("ignore")
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.4.0.post1
Data loading#
The original CITE-seq data (i.e. paired gene expression and surface protein adundance) contains 90,261 bone marrow mononuclear cells. This CITE-seq dataset was generated at 4 different sites introducing some batch effect. After the quality control (QC) performed by the authors, the data contains measurements from 13,953 genes and 134 proteins.
The multiome data (i.e. paired gene expression and chromatin openness) contains 69249 cells from the same donors as the CITE-seq dataset. It also was generated at the same 4 sites. After QC, the data has 13,431 genes and 116,490 peaks.
In this tutorial, we provide subsetted and preprocessed data. Both datasets were randomly subsampled to 20,000 cells each. We also selected 2,000 highly variable genes and 20,000 highly variable pe We use raw RNA counts, centered log-ratio (CLR) normalized data for ADT and log-normalized data for ATAC.
rna_cite_path = "rna_cite.h5ad"
rna_multiome_path = "rna_multiome.h5ad"
atac_path = "atac.h5ad"
adt_path = "adt.h5ad"
try:
rna_cite = sc.read_h5ad(rna_cite_path)
except OSError:
import gdown
gdown.download("https://drive.google.com/uc?export=download&id=1Qzc51EtK0eF5Gi62ucChXHcUxU99Eg-D")
rna_cite = sc.read_h5ad(rna_cite_path)
rna_cite
AnnData object with n_obs × n_vars = 20000 × 2000
obs: 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'cell_type', 'batch', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker'
var: 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'hvg', 'log1p'
try:
atac = sc.read_h5ad(atac_path)
except OSError:
import gdown
gdown.download("https://drive.google.com/uc?export=download&id=17RuUsgKZN41MMfDo6C_oEN4CYWJKnL8H")
atac = sc.read_h5ad(atac_path)
atac
AnnData object with n_obs × n_vars = 20000 × 20000
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker'
var: 'feature_types', 'gene_id', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'ATAC_gene_activity_var_names', 'dataset_id', 'genome', 'hvg', 'log1p', 'organism'
try:
adt = sc.read_h5ad(adt_path)
except OSError:
import gdown
gdown.download("https://drive.google.com/uc?export=download&id=1mlpwwALGDpCCxZS2-laApOEdoYksPhFf")
adt = sc.read_h5ad(adt_path)
adt
AnnData object with n_obs × n_vars = 20000 × 134
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id'
uns: 'dataset_id', 'genome', 'organism'
try:
rna_multiome = sc.read_h5ad(rna_multiome_path)
except OSError:
import gdown
gdown.download("https://drive.google.com/uc?export=download&id=1W1oLSwB9IXL8OTVoxFopn2kqtOfdbXzE")
rna_multiome = sc.read_h5ad(rna_multiome_path)
rna_multiome
AnnData object with n_obs × n_vars = 20000 × 2000
obs: 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'cell_type', 'batch', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker'
var: 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'hvg', 'log1p'
Cell type harmonization#
Since the cell type annotation was performed separately for the CITE-seq and multiome datasets, we perform minimal label harmonization.
celltype_path = "cellttype_harmonize.json"
try:
with open("cellttype_harmonize.json") as f:
harmonized_celltypes = json.load(f)
except OSError:
import gdown
gdown.download("https://drive.google.com/uc?export=download&id=1dWT5PbGqf7I3GSjYLNXJRwdV6GJoQYL4")
with open("cellttype_harmonize.json") as f:
harmonized_celltypes = json.load(f)
harmonized_celltypes.keys()
dict_keys(['cite_ct_l1_map', 'cite_ct_l2_map', 'multi_ct_l1_map', 'multi_ct_l2_map'])
rna_cite.obs["l1_cell_type"] = rna_cite.obs["cell_type"].map(harmonized_celltypes["cite_ct_l1_map"])
rna_cite.obs["l2_cell_type"] = rna_cite.obs["cell_type"].map(harmonized_celltypes["cite_ct_l2_map"])
adt.obs["l1_cell_type"] = adt.obs["cell_type"].map(harmonized_celltypes["cite_ct_l1_map"])
adt.obs["l2_cell_type"] = adt.obs["cell_type"].map(harmonized_celltypes["cite_ct_l2_map"])
rna_multiome.obs["l1_cell_type"] = rna_multiome.obs["cell_type"].map(harmonized_celltypes["multi_ct_l1_map"])
rna_multiome.obs["l2_cell_type"] = rna_multiome.obs["cell_type"].map(harmonized_celltypes["multi_ct_l2_map"])
atac.obs["l1_cell_type"] = atac.obs["cell_type"].map(harmonized_celltypes["multi_ct_l1_map"])
atac.obs["l2_cell_type"] = atac.obs["cell_type"].map(harmonized_celltypes["multi_ct_l2_map"])
Data setup#
Next, we concatenate all the data into one AnnData object specifying how the data is paired and which layers to use. Each sublist in the adatas should correspond to one modality, and the order in each sublist indicates which objects are paired. In the example below, the first sublist corresponds to RNA, the second – to ATAC, and the last – to ADT. We pair rna_cite and adt objects by putting them at the first objects in the sublists, and rna_multiome and atac – as socend. The missing measurements need to be filled with None.
Since we already have the raw counts in .X for rna objects, and normalized data for adt and atac, we don’t need to specify the layers parameter.
rna_cite.X.data, rna_multiome.X.data
(array([ 2., 38., 6., ..., 2., 1., 1.], dtype=float32),
array([1., 2., 1., ..., 3., 1., 1.], dtype=float32))
adt.X.data, atac.X.data
(array([0.38965088, 0.669331 , 0.669331 , ..., 1.054183 , 0.2851298 ,
1.4059474 ], dtype=float32),
array([1.7362776, 1.7362776, 1.7362776, ..., 1.939339 , 1.939339 ,
1.939339 ], dtype=float32))
adata = mtg.data.organize_multimodal_anndatas(
adatas=[[rna_cite, rna_multiome], [None, atac], [adt, None]],
layers=[[None, None], [None, None], [None, None]],
)
adata
AnnData object with n_obs × n_vars = 40000 × 22134
obs: 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'cell_type', 'batch', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'l1_cell_type', 'l2_cell_type', 'group', 'ADT_iso_count', 'ADT_n_antibodies_by_counts', 'ADT_pseudotime_order', 'ADT_total_counts', 'GEX_n_genes_by_counts', 'is_train', 'ATAC_atac_fragments', 'ATAC_blacklist_fraction', 'ATAC_nCount_peaks', 'ATAC_nucleosome_signal', 'ATAC_pseudotime_order', 'ATAC_reads_in_peaks_frac', 'GEX_n_counts', 'GEX_n_genes'
var: 'modality'
uns: 'modality_lengths'
The data comes from 4 different sites, so we select one of the sites as the query.
query = adata[adata.obs["Site"] == "site1"].copy()
adata = adata[adata.obs["Site"] != "site1"].copy()
(adata, query)
(AnnData object with n_obs × n_vars = 31521 × 22134
obs: 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'cell_type', 'batch', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'l1_cell_type', 'l2_cell_type', 'group', 'ADT_iso_count', 'ADT_n_antibodies_by_counts', 'ADT_pseudotime_order', 'ADT_total_counts', 'GEX_n_genes_by_counts', 'is_train', 'ATAC_atac_fragments', 'ATAC_blacklist_fraction', 'ATAC_nCount_peaks', 'ATAC_nucleosome_signal', 'ATAC_pseudotime_order', 'ATAC_reads_in_peaks_frac', 'GEX_n_counts', 'GEX_n_genes'
var: 'modality'
uns: 'modality_lengths',
AnnData object with n_obs × n_vars = 8479 × 22134
obs: 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'cell_type', 'batch', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'l1_cell_type', 'l2_cell_type', 'group', 'ADT_iso_count', 'ADT_n_antibodies_by_counts', 'ADT_pseudotime_order', 'ADT_total_counts', 'GEX_n_genes_by_counts', 'is_train', 'ATAC_atac_fragments', 'ATAC_blacklist_fraction', 'ATAC_nCount_peaks', 'ATAC_nucleosome_signal', 'ATAC_pseudotime_order', 'ATAC_reads_in_peaks_frac', 'GEX_n_counts', 'GEX_n_genes'
var: 'modality'
uns: 'modality_lengths')
Next, we register covariates that the model will correct for in the latent space. We need to pass the length of the RNA data to calculate the library size.
rna_indices_end = rna_cite.shape[1]
# to free memory
del rna_cite, rna_multiome, atac, adt
mtg.model.MultiVAE.setup_anndata(
adata,
categorical_covariate_keys=["Modality", "Samplename"],
rna_indices_end=rna_indices_end,
)
Model setup and training#
We specify integrate_on = 'Modality' to make sure that joint distributions for CITE-seq and multiome data are well aligned. We also need to specify alignment_type="marginal" and modality_alignment = 'MMD' parameters to allow for unimodal reference mapping later. This ensures that the distributions of the CITE-seq and multiome data are aligned in the latent space as well as the unimodal marginal distributions.
The integration coefficient ('integ') in loss_coefs usually requires some fine-tuning and is dependent on the dataset.
If using raw counts for RNA-seq, specify NB loss, if normalized counts, use MSE loss. For ATAC and ADT, we use normalized counts and MSE loss.
vae = mtg.model.MultiVAE(
adata,
losses=["nb", "mse", "mse"],
loss_coefs={
"integ": 500,
},
integrate_on="Modality",
alignment_type="marginal",
modality_alignment="MMD",
)
The user can specify the number of epochs, learning rate and batch size by setting max_epochs, lr and batch_size, respectively. Please refer to the API for more details.
vae.train()
Monitored metric reconstruction_loss_validation did not improve in the last 10 records. Best score: 1038.537. Signaling Trainer to stop.
Visualizing the latent space#
Next, we retrieve the learned latent representation; it is automatically saved in adata.obsm['X_multigrate'].
vae.get_model_output()
adata
AnnData object with n_obs × n_vars = 31521 × 22134
obs: 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'cell_type', 'batch', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'l1_cell_type', 'l2_cell_type', 'group', 'ADT_iso_count', 'ADT_n_antibodies_by_counts', 'ADT_pseudotime_order', 'ADT_total_counts', 'GEX_n_genes_by_counts', 'is_train', 'ATAC_atac_fragments', 'ATAC_blacklist_fraction', 'ATAC_nCount_peaks', 'ATAC_nucleosome_signal', 'ATAC_pseudotime_order', 'ATAC_reads_in_peaks_frac', 'GEX_n_counts', 'GEX_n_genes', 'size_factors', '_scvi_batch'
var: 'modality'
uns: 'modality_lengths', '_scvi_uuid', '_scvi_manager_uuid'
obsm: '_scvi_extra_categorical_covs', 'X_multigrate'
Finally, we visualize the latent space.
sc.pp.neighbors(adata, use_rep="X_multigrate")
sc.tl.umap(adata)
sc.pl.umap(adata, color=["l1_cell_type", "l2_cell_type", "Modality", "Samplename"], ncols=1, frameon=False)

Preparing the query#
Multigrate is equipped with scArches approach [LNL+22] to map new query data onto existing references. Since we have three modalities in the reference, we can map multimodal and unimodal queries (even when we did not have any unimodal data in the reference).
Let’s imitate unimodal queries by masking with zeros the RNA part of one multiome sample and the ADT part of one CITE-seq samples in the query. We get one ATAC-only query sample and one RNA-only query sample respectively.
np.unique(query.obs["Samplename"])
array(['site1_donor1_cite', 'site1_donor1_multiome', 'site1_donor2_cite',
'site1_donor2_multiome', 'site1_donor3_cite',
'site1_donor3_multiome'], dtype=object)
idx_atac_query = query.obs["Samplename"] == "site1_donor1_multiome"
idx_scrna_query = query.obs["Samplename"] == "site1_donor1_cite"
idx_mutiome_query = query.obs["Samplename"].isin(["site1_donor2_multiome", "site1_donor3_multiome"])
idx_cite_query = query.obs["Samplename"].isin(["site1_donor2_cite", "site1_donor3_cite"])
np.sum(idx_atac_query), np.sum(idx_scrna_query), np.sum(idx_mutiome_query), np.sum(idx_cite_query)
(1751, 1140, 3161, 2427)
query[idx_atac_query, :rna_indices_end].X = 0
query[idx_scrna_query, rna_indices_end:].X = 0
Next, we initialize a new model taking the weights from the reference model but add new weights for the query batches, i.e. samples, following the scArches approach [LNL+22].
new_vae = mtg.model.MultiVAE.load_query_data(query, vae)
Next, we fine-tune the newly added weights to optimize the reconstruction of the query data. We set weight_decay to zero to make sure that the rest of the weights in the model will not be changed.
new_vae.train(weight_decay=0)
Now, we obtain the latent representation of the query and visualize both the reference and the query together.
new_vae.get_model_output(query)
query
AnnData object with n_obs × n_vars = 8479 × 22134
obs: 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'cell_type', 'batch', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'l1_cell_type', 'l2_cell_type', 'group', 'ADT_iso_count', 'ADT_n_antibodies_by_counts', 'ADT_pseudotime_order', 'ADT_total_counts', 'GEX_n_genes_by_counts', 'is_train', 'ATAC_atac_fragments', 'ATAC_blacklist_fraction', 'ATAC_nCount_peaks', 'ATAC_nucleosome_signal', 'ATAC_pseudotime_order', 'ATAC_reads_in_peaks_frac', 'GEX_n_counts', 'GEX_n_genes', 'size_factors', '_scvi_batch'
var: 'modality'
uns: 'modality_lengths', '_scvi_uuid', '_scvi_manager_uuid'
obsm: '_scvi_extra_categorical_covs', 'X_multigrate'
adata.obs["reference"] = "reference"
query.obs["reference"] = "query"
adata.obs["type_of_query"] = "reference"
query.obs.loc[idx_atac_query, "type_of_query"] = "ATAC query"
query.obs.loc[idx_scrna_query, "type_of_query"] = "scRNA query"
query.obs.loc[idx_mutiome_query, "type_of_query"] = "multiome query"
query.obs.loc[idx_cite_query, "type_of_query"] = "CITE-seq query"
adata_both = ad.concat([adata, query])
sc.pp.neighbors(adata_both, use_rep="X_multigrate")
sc.tl.umap(adata_both)
sc.pl.umap(
adata_both, color=["l1_cell_type", "l2_cell_type", "reference", "Modality", "Samplename"], ncols=1, frameon=False
)
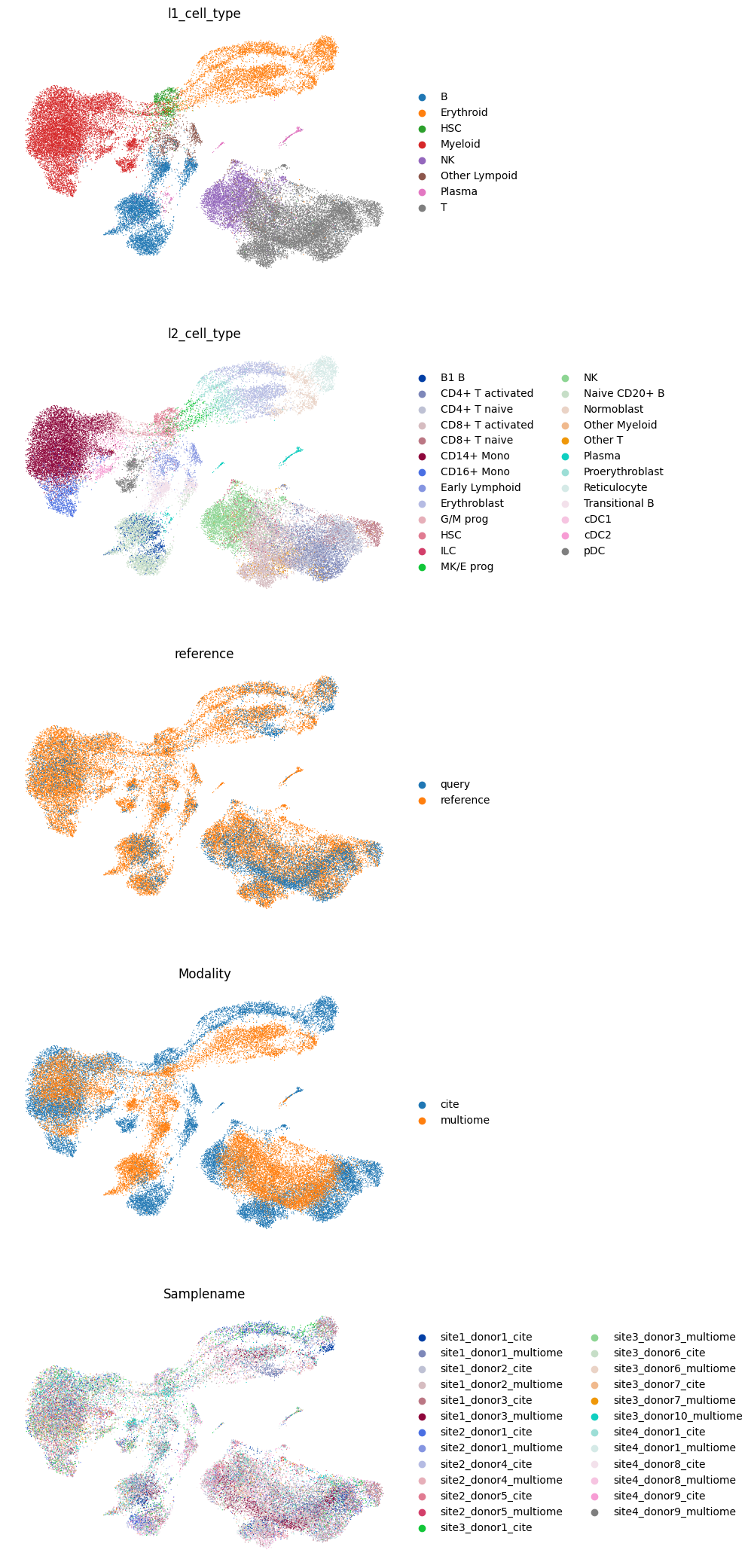
We also can take a look at separate multimodal and unimodal queries.
sc.pl.umap(adata_both, color="type_of_query", ncols=1, frameon=False, groups=["CITE-seq query"])

sc.pl.umap(adata_both, color="type_of_query", ncols=1, frameon=False, groups=["multiome query"])

sc.pl.umap(adata_both, color="type_of_query", ncols=1, frameon=False, groups=["scRNA query"])

sc.pl.umap(adata_both, color="type_of_query", ncols=1, frameon=False, groups=["ATAC query"])
